![بیماری کاناوان [canavan] چیست + علائم، علت و درمان کاناوان](https://salamdonya.com/assets/images/58/5888vzq79.jpg)
بیماری کاناوان از بیماری های ژنتیکی می باشد که مربوط به بافت مغز است، در این مطلب از سلام دنیا درباره بیماری کاناوان چیست، علائم و نشانه ها بیماری کاناوان، علت وجود بیماری کاناوان، اختلالات مرتبط، روش تشخیص و درمان بیماری کاناوان صحبت خواهیم کرد. با ما همراه باشید.
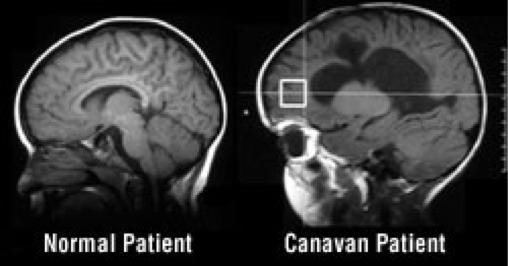
بیماری کاناوان (canavan) چیست
بیماری کاناوان یک اختلال عصبی ژنتیکی نادر است که با انحطاط اسفنجی ماده سفید در مغز مشخص می شود. نوزادان مبتلا ممکن است در بدو تولد طبیعی به نظر برسند، اما معمولاً علائم را بین 3 تا 6 ماهگی نشان می دهند. علائم ممکن است شامل:
- سر بزرگ (ماکروسفالی)
- عدم کنترل سر
- کاهش شدید تون عضلانی که منجر به “شکستگی” می شود
- تاخیر در رسیدن به نقاط عطف رشد مانند نشستن و راه رفتن که به صورت مستقل باشد.
حتما بخوانید: هیدروسفالی یا آب آوردن مغز چیست
اکثر کودکان مبتلا تا 10 سالگی دچار عوارض تهدید کننده زندگی می شوند. بیماری کاناوان به دلیل جهش در ژن آسپارتواسیلاز (ASPA) رخ میدهد که بر تجزیه (متابولیسم) N-acetylaspartic اسید (NNA) تأثیر میگذارد. به عنوان یک بیماری اتوزومال مغلوب به ارث می رسد.

چه کسانی به بیماری کاناوان مبتلا می شوند
بیماری کاناوان به گروهی از اختلالات معروف به لوکودیستروفی تعلق دارد. لوکودیستروفی ها گروهی از اختلالات نادر، پیش رونده، متابولیک و ژنتیکی هستند که می توانند مغز، نخاع و اغلب اعصاب خارج از سیستم عصبی مرکزی (اعصاب محیطی) را تحت تاثیر قرار دهند. هر نوع لوکودیستروفی ناشی از یک ناهنجاری است که بر یک ژن خاص تأثیر می گذارد که منجر به رشد غیرطبیعی یکی از حداقل 10 ماده شیمیایی مختلف می شود که ماده سفید مغز را تشکیل می دهند. ماده سفید بافتی است که از رشته های عصبی تشکیل شده است. بسیاری از این رشته های عصبی توسط مجموعه ای از چربی ها (لیپیدها) و پروتئین هایی به نام میلین پوشیده شده اند. میلین، که در مجموع ممکن است به عنوان غلاف میلین شناخته شود، از رشته های عصبی محافظت می کند، به عنوان یک عایق عمل می کند و سرعت انتقال سیگنال های عصبی را افزایش می دهد.
علائم و نشانه ها بیماری کاناوان
علائم و پیشرفت بیماری کاناوان در هر فردمتفاوت است. این اختلال معمولاً بین 3 تا 6 ماهگی آشکار میشود و علائم اولیه معمولاً شامل:
- کنترل بسیار ضعیف سر، بزرگی غیرطبیعی سر (ماکروسفالی) و کاهش شدید تون عضلانی (هیپوتونی) است که منجر به شل شدن میشود.
- نوزادان مبتلا ممکن است به طور کلی بی پاسخ (بی تفاوت)، بی حال یا تحریک پذیر باشند. برخی از نوزادان ممکن است دچار مشکل در بلع (دیسفاژی) شوند که به مشکلات تغذیه کمک می کند.
- نوزادان مبتلا همچنین در رسیدن به نقاط عطف رشد (مانند نشستن یا ایستادن بدون کمک) تاخیر نشان میدهند و اکثر آنها هرگز مستقل راه نمیروند.
- از دست دادن تدریجی توانایی هایی که مستلزم هماهنگی فعالیت ذهنی و عضلانی است (رگرسیون روانی حرکتی) و عقب ماندگی ذهنی نیز در دوران نوزادی آشکار می شود.
- بیشتر نوزادان مبتلا یاد می گیرند که لبخند بزنند، بخندند، سر خود را بالا ببرند و تعامل اجتماعی داشته باشند.
علائم دیگری که کودکان مبتلا به بیماری کاناوان را تحت تاثیر قرار می دهد شامل:
- تشنج
حتما بخوانید: انواع تشنج چیست و چگونه درمان میشود
- اختلالات خواب
- مشکلات تغذیه
- نارسایی بینی
- برگشت اسید از معده به مری (رفلاکس) که گاهی با استفراغ همراه است
- زوال اعصاب چشم (اعصاب بینایی) است
- انتقال تکانه ها از غشای غنی از اعصاب پوشاننده چشم (شبکیه) به مغز (آتروفی بینایی).(آتروفی بینایی ممکن است باعث کاهش پاسخ بینایی شود) در بیشتر موارد، شنوایی تحت تأثیر قرار نمی گیرد، اما کاهش شنوایی ممکن است رخ دهد.
علائم بیماری کاناوان در سن بالاتر نوزادان:
با افزایش سن نوزادان مبتلا، هیپوتونی ممکن است در نهایت به اسپاسم تبدیل شود، وضعیتی که با اسپاسم غیرارادی عضلانی مشخص می شود که منجر به حرکات آهسته و سفت پاها می شود. افراد مبتلا ممکن است در نهایت امتداد و چرخش سفت و سخت کنترل نشده بازوها، پاها، انگشتان دست و پا (سفتی دسربراتور) یا فلج را نشان دهند. بیماری کاناوان در نهایت پیشرفت می کند و باعث ایجاد عوارض تهدید کننده زندگی می شود. با این حال، شدت و پیشرفت بیماری متفاوت است. برخی از افراد در دوران نوزادی دچار عوارض تهدید کننده زندگی می شوند. دیگران فراتر از سنین نوجوانی خود زندگی می کنند.
در چند سال اخیر، نوع خفیف بیماری کاناوان با جهش های مشخصه ژن ASPA و تنها افزایش اندکی NAA در ادرار شناسایی شده است. این کودکان ممکن است فقط کمی تاخیر داشته باشند، می توانند یاد بگیرند و به مدرسه بروند. سر ممکن است تا حدودی بزرگ شده باشد، اما تغییرات ماده سفید معمولی مرتبط با Canavan ممکن است وجود نداشته باشد. اگاهی از قبل مطمئناً بسیار بهتر است.
علت بروز بیماری کاناوان
بیماری کاناوان در اثر اختلال یا تغییر (جهش) در ژن آسپارتواسیلاز (ASPA) ایجاد می شود. این جهش به عنوان یک صفت اتوزومال مغلوب به ارث می رسد. بیماریهای ژنتیکی با ترکیب ژنهای یک صفت خاص که روی کروموزومهای دریافت شده از پدر و مادر هستند، تعیین میشوند.
اختلالات ژنتیکی مغلوب زمانی اتفاق میافتد که یک فرد ژن غیر طبیعی یکسانی را برای یک صفت از هر یک از والدین به ارث میبرد. اگر فردی یک ژن طبیعی و یک ژن برای بیماری دریافت کند، فرد ناقل بیماری خواهد بود، اما معمولاً علائمی از خود نشان نخواهد داد. خطر این که دو والدین ناقل هر دو ژن معیوب را منتقل کنند و در نتیجه فرزندی مبتلا داشته باشند در هر بارداری 25 درصد است. خطر داشتن فرزندی که مانند والدین ناقل است در هر بارداری 50 درصد است. شانس اینکه کودک از هر دو والدین ژن طبیعی دریافت کند و از نظر ژنتیکی برای آن صفت خاص طبیعی باشد 25 درصد است. این خطر برای مردان و زنان یکسان است.
ژن معیوب مسئول بیماری Canavan به کروموزوم 17 (17pter-p13) نگاشت شده است. کروموزوم هایی که در هسته سلول های انسانی وجود دارند، حامل اطلاعات ژنتیکی هر فرد هستند. سلول های بدن انسان به طور معمول دارای 46 کروموزوم هستند. جفت کروموزوم های انسان از 1 تا 22 شماره گذاری می شوند و کروموزوم های جنسی X و Y مشخص می شوند. مردان دارای یک کروموزوم X و Y و ماده ها دارای دو کروموزوم X هستند. هر کروموزوم دارای یک بازوی کوتاه به نام “p” و یک بازوی بلند با نام “q” است. کروموزوم ها بیشتر به نوارهای متعددی تقسیم می شوند که شماره گذاری می شوند. به عنوان مثال، “کروموزوم 11p13” به نوار 13 در بازوی کوتاه کروموزوم 11 اشاره دارد. نوارهای شماره گذاری شده محل هزاران ژن موجود در هر کروموزوم را مشخص می کنند.
ASPA حاوی دستورالعمل هایی برای توسعه (رمزگذاری) آسپارتواسیلاز، آنزیمی است که N-acetylaspartic acid (NAA) را تجزیه می کند (متابولیزه می کند). NAA ترکیبی است که محققان معتقدند نقش حیاتی در حفظ ماده سفید مغز دارد. کمبود یا غیر فعال بودن آسپارتواسیلاز منجر به تجمع NAA در بافت مغز می شود. علائم بیماری Canavan ناشی از آسیب به ماده سفید ناشی از سطوح بالای غیر طبیعی NAA است.
جمعیت های تحت تاثیر بیماری کاناوان
بیماری کاناوان مردان و زنان را به تعداد مساوی درگیر می کند. این بیماری همه گروه های قومی را تحت تاثیر قرار می دهد، اما با فراوانی بیشتری در افراد یهودی تبار اشکنازی رخ می دهد. در این جمعیت، فراوانی ناقل از هر 40 تا 58 نفر یک نفر تخمین زده می شود. خطر تولد کودک مبتلا از والدین یهودی اشکنازی بین 1 تا 6400 و 1 در 13456 است. فرکانس حامل در سایر جمعیت ها مشخص نیست، اما به احتمال زیاد بسیار کمتر است. بروز کلی بیماری کاناوان در جمعیت عمومی ناشناخته است.
حتما بخوانید: علت بیماری آدیسون چیست
اختلالات مرتبط بیماری کاناوان
علائم اختلالات زیر می تواند مشابه علائم بیماری کاناوان باشد. مقایسه ممکن است برای تشخیص افتراقی مفید باشد:
1. لوکودیستروفی ها
گروهی از بیماری های بسیار نادر، پیش رونده، متابولیک و ژنتیکی هستند که مغز، نخاع و اغلب اعصاب محیطی را تحت تاثیر قرار می دهند. هر نوع لوکودیستروفی ناشی از یک ناهنجاری ژنی خاص است که منجر به رشد غیرطبیعی یکی از حداقل 10 ماده شیمیایی مختلف می شود که ماده سفید (غلاف میلین) مغز را تشکیل می دهد. غلاف میلین پوشش محافظ عصب است و اعصاب بدون آن نمی توانند به طور طبیعی کار کنند. هر نوع لوکودیستروفی بخش متفاوتی از غلاف میلین را تحت تاثیر قرار می دهد و منجر به طیف وسیعی از مشکلات عصبی می شود.
2. لوکودیستروفی متاکروماتیک
شایع ترین شکل لوکودیستروفی، یک اختلال نورومتابولیک ارثی نادر است که ماده سفید مغز را تحت تاثیر قرار می دهد (لوکوآنسفالوپاتی). مشخصه آن تجمع یک ماده چرب به نام سولفاتید (اسفنگولیپید) در مغز و سایر نواحی بدن (یعنی کبد، کیسه صفرا، کلیه ها و/یا طحال) است. پوشش محافظ چربی روی رشته های عصبی (میلین) به دلیل تجمع سولفاتید از ناحیه سیستم عصبی مرکزی (CNS) از بین می رود. علائم لوکودیستروفی متاکروماتیک ممکن است شامل تشنج،تغییرات شخصیتی، اسپاستیسیتی، زوال عقل پیشرونده، اختلالات حرکتی در حال پیشرفت به فلج و/یا اختلال بینایی منجر به کوری باشد. لوکودیستروفی متاکروماتیک به عنوان یک صفت اتوزومال مغلوب به ارث می رسد.
3. بیماری تای ساکس
یک اختلال نادر و تخریبکننده عصبی است که در آن کمبود آنزیم (هگزوزامینیداز A) منجر به تجمع بیش از حد برخی چربیها (لیپیدها) معروف به گانگلیوزیدها در مغز و سلولهای عصبی میشود. این تجمع غیر طبیعی گانگلیوزیدها منجر به اختلال پیشرونده سیستم عصبی مرکزی می شود. این اختلال به عنوان یک بیماری ذخیره لیزوزومی طبقه بندی می شود. لیزوزوم ها واحدهای گوارشی اصلی در سلول ها هستند. آنزیمهای درون لیزوزومها، مواد مغذی، از جمله کربوهیدراتهای پیچیده و چربیها را تجزیه یا «هضم» میکنند.
علائم مرتبط با بیماری تای ساکس ممکن است شامل پاسخ مبهوت کننده مبالغه آمیز به صداهای ناگهانی، بی حالی، از دست دادن مهارت های قبلی اکتسابی (به عنوان مثال، رگرسیون روانی حرکتی)، و کاهش شدید تون عضلانی (هیپوتونی) باشد. با پیشرفت بیماری، نوزادان و کودکان مبتلا ممکن است لکه های قرمز گیلاسی در لایه میانی چشم، از دست دادن تدریجی بینایی و ناشنوایی، افزایش سفتی عضلات و محدودیت حرکات (اسپاستیسیته)، فلج نهایی، اختلالات الکتریکی کنترل نشده در مغز (تشنج) و بدتر شدن فرآیندهای شناختی (زوال عقل). بیماری تای ساکس به عنوان یک صفت اتوزومال مغلوب به ارث می رسد.
4. برخی از اختلالات میتوکندریایی
مانند بیماری لی، ممکن است با دژنراسیون اسفنجی سیستم عصبی مرکزی همراه باشد. اختلالات میتوکندری با جهش هایی مشخص می شود که بر بخش هایی از سلول که انرژی آزاد می کنند (میتوکندری) تأثیر می گذارد. بیماری های میتوکندری اغلب توانایی سلول های آسیب دیده را برای تجزیه غذا و اکسیژن و تولید انرژی مختل می کند. در بیشتر اختلالات میتوکندریایی، تعداد غیرطبیعی زیادی از میتوکندری های معیوب در سلول های بدن وجود دارد. بیماری های میتوکندری اغلب بیش از یک سیستم اندام بدن را تحت تأثیر قرار می دهند.
تشخیص بیماری کاناوان
تشخیص بیماری کاناوان ممکن است در نوزادانی با یافته های مشخصه این اختلال (مانند کنترل ضعیف سر، ماکروسفالی و غیره) مشکوک باشد. تشخیص ممکن است با ارزیابی بالینی کامل، شرح حال دقیق بیمار و انواع آزمایشات تخصصی تایید شود. چنین آزمایشاتی ممکن است شامل کروماتوگرافی گازی – طیف سنجی جرمی باشد، دستگاهی که می تواند سطوح بالای NAA را در ادرار تشخیص دهد. سطوح بالای NAA همچنین در خون و مایع مغزی نخاعی (CSF) قابل تشخیص است. بررسی برخی سلول های بافت همبند از پوست (فیبروبلاست های کشت شده) می تواند کمبود آنزیم آسپارتواسیلاز را نشان دهد. فعالیت آسپارتواسیلاز نیز در گلبول های سفید وجود ندارد.
تشخیص قبل از تولد بیماری Canavan از طریق آمنیوسنتز با اندازه گیری سطح NAA در مایعی که جنین در حال رشد (مایع آمنیوتیک) را در هفته 16-18 بارداری احاطه کرده است، در دسترس است. اگر هر دو والدین جهشهای ژن ASPA را شناختهاند، تشخیص قبل از تولد با استفاده از نمونهگیری از پرزهای کوریونی (CVS) در دسترس است که در آن نمونهای از سلولهای جفت در هفته 10-12 بارداری برای تجزیه و تحلیل جهش برداشته میشود.
درمان بیماری کاناوان
درمان های استاندارد
درمان بیماری کاناوان به سمت علائم خاصی است که در هر فرد آشکار است. مراقبت های حمایتی ممکن است برخی از ناراحتی ها را کاهش دهد. فیزیوتراپی و مداخله زودهنگام ممکن است به ترتیب به بهبود وضعیت وضعیت بدن و مهارت های ارتباطی کمک کنند. در صورت بروز مشکلات در بلع، لوله های تغذیه ممکن است برای اطمینان از تغذیه مناسب و هیدراتاسیون مفید باشد. تشنج ممکن است با داروهای ضد تشنج (ضد تشنج) درمان شود. مشاوره ژنتیک و آزمایش ناقل برای خانواده هایی که این بیماری در آنها رخ می دهد مفید خواهد بود.

درمان های تحقیقاتی
محققان در حال مطالعه ژن درمانی برای درمان کودکان مبتلا به بیماری Canavan هستند. در ژن درمانی، نسخه های سالم ژن معیوب ASPA در مغز کودکان مبتلا قرار می گیرد. سپس این ژن ها آنزیم آسپارتواسیلاز مورد نیاز برای تجزیه NAA را تولید می کنند. کودکانی که تحت درمان با ژن درمانی قرار گرفته اند، بهبود قابل توجهی در علائم نشان داده اند. تحقیقات بیشتری برای تعیین ایمنی و اثربخشی طولانی مدت ژن درمانی به عنوان یک درمان بالقوه برای افراد مبتلا به بیماری کاناوان ضروری است.
سخن آخر
با پیشرفت علم زنان و مردان قبل از ازدواج و پیش از اقدام به بارداری می توانند از بروز تولد نوزادان با مشکلات ژنتیکی جلوگیری کنند.
دیدگاه ها